| 2017-03-09 09:35:30 -0600 | answered a question | Bam file Error code 1 Ion Torrent data Hi Rune, I am sorry you are having issues visualizing your BAM files. That error message is usually caused by permissions issues to the directory where the BAM file is saved. Can you verify that you have read/write permissions to the directory? Alternatively, if you copy the BAM file to a local drive on your computer is GenomeBrowse able to read the file? Let us know if you continue to have issues. Thanks, Jami Bartole Golden Helix, Inc. |
| 2017-02-13 15:56:55 -0600 | answered a question | Arabidopsis TAIR9 reference genome Hi Shaun, To add a new genome assembly to GenomeBrowse requires the nucleotide sequence in FASTA format. Once you have the appropriate files you can go to Add then select Convert from the lower left corner of the Data Source Library. This will open up the Convert Wizard so you can add in your custom data source. You can find a description of converting the FASTA file(s) in our manual at the following link. http://doc.goldenhelix.com/GenomeBrowse/latest/gbmanual/convertsourcewizard.html#converting-a-fasta-file Once the file is converted to a usable format you can select the assembly from dropdown list.
Then select the Add button to choose the new file from your Local annotations folder. Let me know if you have any further questions. Thanks, Jami... |
| 2016-11-15 15:51:34 -0600 | answered a question | Link to S. pombe genome is broken Hi David, I am sorry you have found a broken link. Can you provide some screenshots of where you see the link and I can take a look at getting it fixed? Custom genomes can be added using a reference sequence FASTA file. Just go to Tools > Manage Data Sources and click the Convert button in the lower left corner. Once the convert wizard appears just load in your FASTA file and follow the prompts to create the file. You can find further details of this process in our manual at the following link. Let me know how I can help you further. Thanks, Jami. Golden Helix, Inc. |
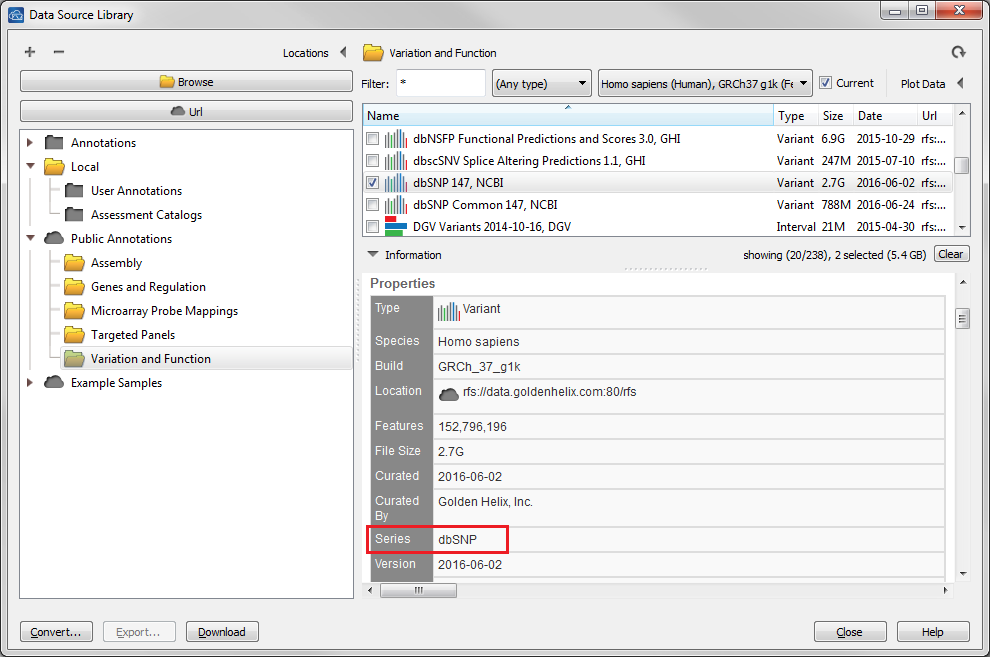
| 2016-10-27 11:11:44 -0600 | answered a question | Which dbSNP source should be used for 23andme raw data? Hi Carl, To fully import your 23andMe variants you will want to select the full dbSNP 147 track and not the common subset as it will contain the most complete list of variants including the rare ones. When you load your 23andMe data into the Convert Wizard it will auto detect the most recent track with "dbSNP" listed as the series name, so it should not even consider the common subsets as the series name is "dbSNPCommon".
Let me know how I can help you further. Thanks, Jami... |
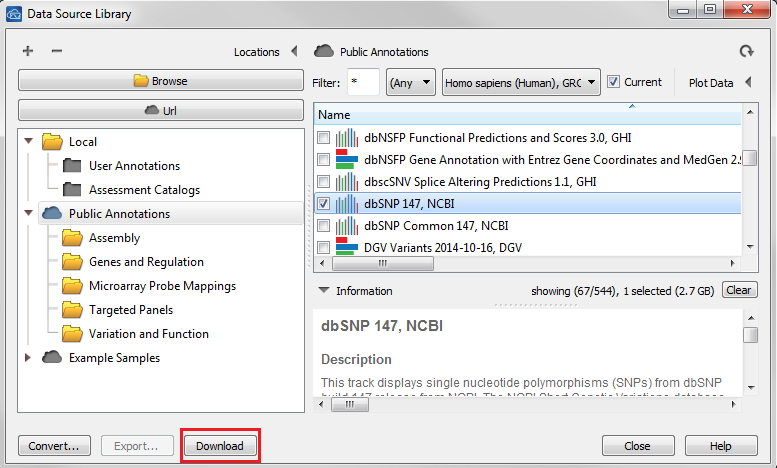
| 2016-10-11 10:22:33 -0600 | answered a question | I would like to convert 23andme data but am stopped by step below? Hi Janet, That message just means that you need a local copy of a dbSNP source downloaded for the import to work correctly. You can do this by going to Tools > Manage Data Source and selecting a dbSNP track from the Public Annotations repository then click Download at the bottom of the window.
Once the download is finished you may need to hit refresh in the upper right corner of the dialog for the track to appear in your Local annotation folder. Once you can see the track in the Local folder then proceed to the Convert Wizard and add your 23andMe data, it should correctly pick-up the newly downloaded SNP track automatically. Let me know if you have any further issues. Thanks, Jami... Golden Helix, Inc. |
| 2016-08-08 14:55:19 -0600 | answered a question | How can I convert GTF to GFF file format? Dear Fee, Unfortunately GenomeBrowse does not have the functionality to convert GTF format to GFF file format. However, the Convert Wizard that is available within the software can directly support conversion of the GTF formatted data. The Convert Wizard can be accessed by going to File > Add then clicking Convert in the lower left corner of the Data Source Library. Once the GTF data is converted to TSF (Golden Helix annotation format) then it can be loaded into the GenomeBrowse plot window for visualization. If you are not looking to visualize the data in GenomeBrowse then there are several third party conversion tools available to get your data in GFF format. Below is one example from the Sequence Ontology group. http://www.sequenceontology.org/cgi-bin/converter.cgi Let me know if you have any further questions. Thanks, Jami... |
| 2016-07-25 16:29:16 -0600 | answered a question | bed12 support? Hi Shuoguo, I am glad you have been enjoying GenomeBrowse. The current BED format supported in GenomeBrowse is focused around displaying "generic" intervals that have just a start, stop and potentially a label and color defined in the data. Does your BED file contain transcript data, if so they we recommend getting the same data available in GTF as GenomeBrowse can easily parse this for visualization of the individual transcripts. Another option is to load the BED file into our Convert Wizard (select File > Add then click Convert in the lower left corner) which will allow you to adjust some of the visualization options of the raw data. If you can provide an example of a plot that shows the data in your expected format that would be most useful in determining where GenomeBrowse can be updated with that feature. Let me know how I can help you further. Thanks, Jami... |
| 2016-07-11 10:36:06 -0600 | answered a question | de novo mutation Dear Chrigi, GenomeBrowse is for visualization purposes only and does not have functionality for identifying de Novo mutations. The tutorials available on our website are designed for our analysis software SNP & Variation Suite (SVS). SVS allows you to perform analysis on data in supported formats, to identify de Novo candidates you will minimally need VCF files for your proband and at least one parent. If you would like to try out SVS for your analysis needs please go to our website at the following link and click the button to request a trial of the software. http://goldenhelix.com/products/SNP_Variation/index.html Please let us know how we can help you further. Thanks, Jami... Golden Helix, Inc. |
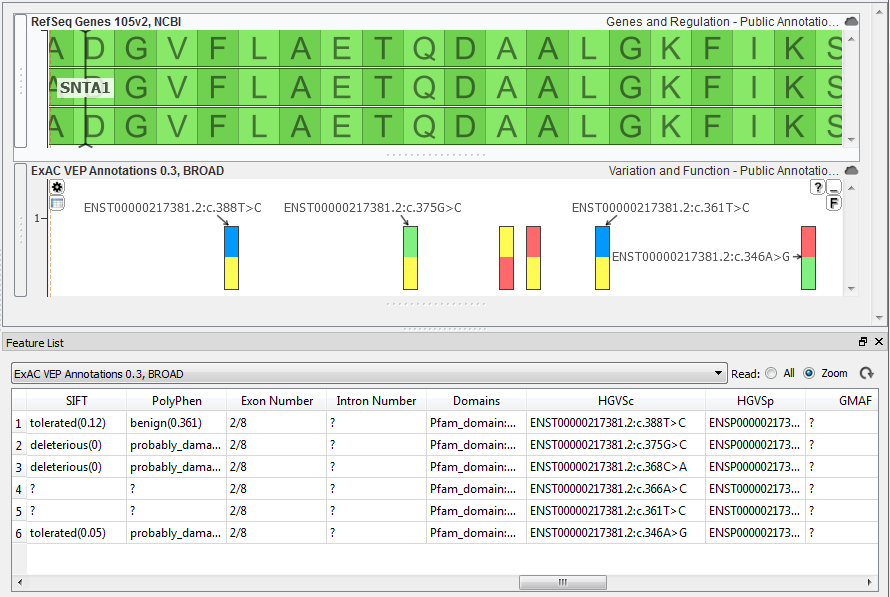
| 2016-05-17 16:40:38 -0600 | answered a question | c. positions Hi Kyle, Visualizations in GenomeBrowse are limited to what is available within the annotation sources or data files that are currently loaded into the window, we do not perform any type of computations on the data. With variant sources there are a few annotation sources that will provide c. notation for the variant. The most comprehensive source is the ExAC VEP Annotations 0.3, BROAD source that is available from the Public Annotations repository (File > Add). If you load this source any variant in your data that is also in the ExAC track will contain this information.
To make the HGVS c. notation the label on the plot just hover your mouse over the ExAC plot and you should see a gear icon appear in the upper left corner, just click the icon to change the labels. Let me know if you have further questions. Thanks, Jami... |
| 2016-05-16 09:32:47 -0600 | answered a question | How can I estimate the linkage disequilibrium from my imputed data set? Hi Mario, I am happy to help with your GenomeBrowse questions. The stand-alone version of GenomeBrowse is for visualization of data only, it does not have the functionality to estimate LD between markers. Linkage Disequilibrium estimations are available in our SNP & Variant Suite (SVS) product. You can see an example of this analysis in our tutorial at the following link. http://doc.goldenhelix.com/SVS/tutorials/ldhaplotypeanalysis/index.html You can request access to SVS by filling out a request on our website at the following link. http://goldenhelix.com/products/SNP_Variation/index.html Let us know if you have any further questions. Thanks, Jami... Golden Helix Support |
| 2016-05-12 15:52:54 -0600 | answered a question | Can't get plot to display Hi Whit, I am sorry you are having issues visualizing your data in GenomeBrowse. With mitochondrial data on the GRCh37 genome you will need to make sure you have the correct reference assembly specified in the drop down at the top of the GenomeBrowse window that matches the chromosome naming convention for this region. In particular, for the hg19 version of the assembly chrM is used to define this region but for the g1k version of the assembly chrMT is used. If you click on the name of your BAM file in the GenomeBrowse plot and then scroll to the bottom of the Console window you will see how this region is identified in your BAM file. Should look similar to the below screenshot.
So you will want to make sure and select the appropriate assembly for your data. If that does not work to fix the visibility can you try deleting the index file (BAI) that should be in the same directory as the BAM file? GenomeBrowse will then try and recompute the file if there are issues with the data you should get some error messages that may help diagnose what is going wrong. Screenshots of any error messages would be most useful. The read depth of the data should not cause this issues, when the number of reads exceeds what can be rendered you will still see those that fit you will just get and error message saying not all reads could be drawn. Let me know how I can help you further. Thanks, Jami... |
| 2016-03-29 09:24:35 -0600 | answered a question | showing read sequence content beyond clipping point Hi Charlie, Visualization of sequencing data is restricted to the information that is provided in the BAM files for your data. Therefore showing content beyond the clipping point is not available since it would not have been included in the alignment output and GenomeBrowse does not support visualization of the raw reads from the FASTQ file. Let me know if you have any further questions. Thanks, Jami... |
| 2016-03-11 16:34:58 -0600 | answered a question | Adding new genome assemblies Hi Sagi, I wanted to let you know that we have added support for the Staphylococcus aureus RF122 genome. You can download the Reference Sequence and Gene tracks by going to File > Add and selecting them from the Public Annotations location. Let me know if you have any issues or questions about the new annotation sources. Thanks, Jami... |
| 2016-03-03 14:09:46 -0600 | answered a question | why I can not load Hi Christian, I am sorry you are having issues loading your BAM files into GenomeBrowse. Can you provide a screenshot of the exact error you are receiving when loading your file? GenomeBrowse has some specific requirements that are necessary to visualize BAM data, in particular a matching reference sequence must be identified for coverage to be computed. To match to a reference sequence the chromosome names and lengths must match exactly to what is reported in the header for the BAM file. Please let me know how I can be of further assistance. Thanks, Jami... |
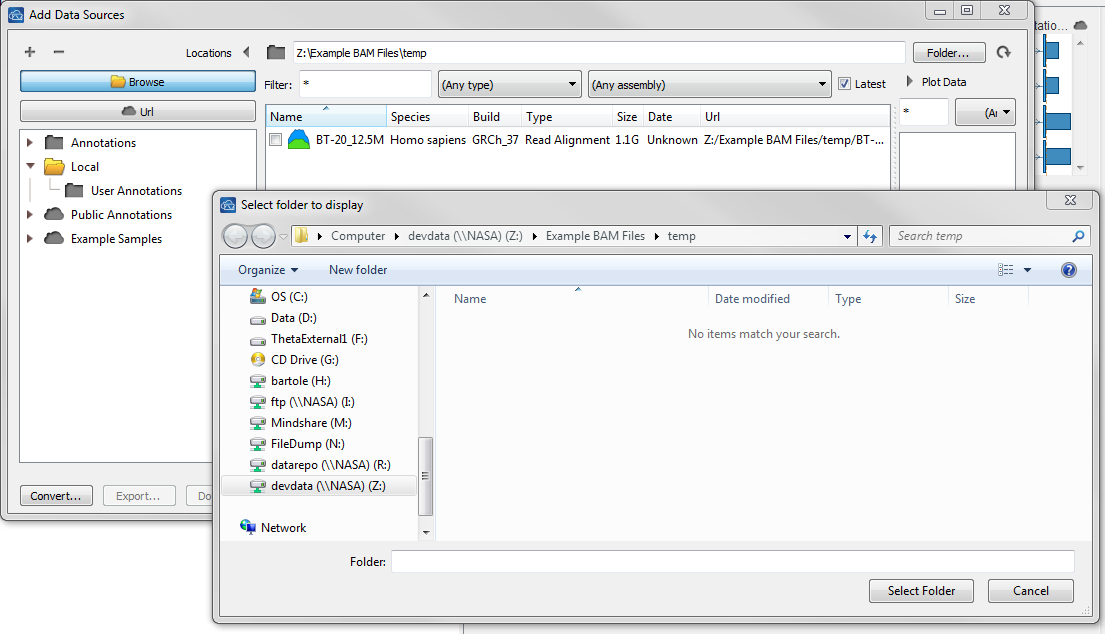
| 2016-02-29 15:10:45 -0600 | answered a question | Why I can not find my files? Hi Christian, I am sorry you are having issues loading your BAM and VCF files into GenomeBrowse. The File > Add option should open the Add Data Sources dialog so you can navigate to the folder where you BAM and VCF files are saved. When you click Browse and then Folder, the files will not be visible in the Folder window if you are using GenomeBrowse on a Windows machine as it will only show existing folders. However, once you click Select Folder where the files are stored you should see them in the Add Data Sources window. Should look similar to the following screenshot.
With Genome Browse you also have the option of dragging your files from a File Explorer window into the plot window, this will directly add them to the plot. If you are still having issues can you provide some screenshots of what you are seeing and I can provide further suggestions. Thanks, Jami... support@goldenhelix.com |
| 2016-01-29 14:57:37 -0600 | answered a question | Can GenomeBrowse read BS-seq BAM files produced by Bismark? Hi Kim, Unfortunately GenomeBrowse can only visualize sorted BAM files. Since your files are not sorted you will not be able to use GenomeBrowse until you can get the files sorted in genomic order. There are several options for sorting your files including SAMTools, which is a free command line program for working with SAM/BAM files. Let me know if you have any further questions. Thanks, Jami... |
| 2016-01-26 17:02:35 -0600 | answered a question | Bug reports and suggestions Hi Ted, We are happy to take any bug reports and feature requests on the site here. Just create a question with your report or suggestions. Thanks, Jami... Senor Field Application Scientist Golden Helix, Inc. |
| 2015-12-31 10:02:58 -0600 | answered a question | Annotation tracks for pan troglodytes Hi Martin, I wanted to let you know that we have been able to add support for the chimp assemblies PanTro3 and PanTro4 to GenomeBrowse. You can download the reference sequence tracks for both assemblies and the gene annotation source for the PanTro4 assembly by going to Add and selecting them from the Public Annotations location. They are listed under builds CSAC2.1.3 and CSAC2.1.4 for the Pan troglodytes species. Let me know if you have any issues or further questions. Thanks, Jami... |
| 2015-12-31 08:56:51 -0600 | commented answer | How do I switch between viewing the negative strand and positive strand in a sequence? Hi Whit, I am not entirely sure about what you are seeing in your GenomeBrowse view. Can you provide a screenshot on the site here or email it to support@goldenhelix.com and I can take a look to see how we can help. Thanks, Jami... |
| 2015-12-30 15:03:53 -0600 | answered a question | How do I switch between viewing the negative strand and positive strand in a sequence? Hi Whit, Are you looking at a reference sequence file or a gene annotation file? Both forward and reverse strand information is available for any reference sequence plot just by increasing the vertical height of the graph. It would look something like the below figure, where the top information is for the forward strand and the bottom information is the reverse strand at this location.
If you are looking at a gene annotation file then the strand for a gene is fixed based on strand field listed in the source of the annotation information.
Any gene listed on the forward strand will be colored blue and any gene on the reverse strand will be green. Let me know if you have any further questions. Thanks, Jami... |
| 2015-10-26 15:28:53 -0600 | answered a question | Fail to launch on Ubuntu 15.04 Hi Roberto, The libraries for GenomeBrowse are compiled on Ubuntu 12.04 so can sometimes have issues with new versions of the operating system. Moving the shipped libraries out of the way so that the system libraries can be used is exactly what we recommend for fixing the error message that you reported. Please let us know if you have any further issues or questions. Thanks, Jami... |
| 2015-10-15 14:46:15 -0600 | answered a question | Adding new genome assemblies Hi Sagi, I am sorry you are having issues adding your genome of interest to GenomeBrowse. Unfortunately the stand-alone version of the software does not directly support converting gene annotations in GFF3 format. As you have seen we only support the GTF format that is based off the specifications determined by Washington State University in St. Louis (http://mblab.wustl.edu/GTF22.html). If you can provide a sample of your converted GTF file I would be happy to take a look to see why the Convert Wizard is having issues. I will also add this genome to the list of genomes to directly support through GenomeBrowse. It usually take a few weeks for them to be added and I will let you know when it can be downloaded from our Public Annotations. Let me know if you have any questions. Thanks, Jami... |
| 2015-10-09 14:37:17 -0600 | answered a question | Does GenomeBrowse display the coding designation for a particular base? Hi Paul, I am happy to help with your GenomeBrowse questions. Just to clarify are you looking for the HGVS C dot notation (ex. NM_001277333.1:c.562G>T) for all variants in your data? If so there are a few tracks that have this information for their listed variants, for example the "COSMIC Mutations Left Aligned 71 v2, GHI" track provides this information in the Mutation CDS field. This is a cancer specific track so only those cancer specific mutations are included. If you are looking to have this information for all of your variants then some type of variant classification algorithm is required to determine the position of the variant within gene regions to create the C dot notation. GenomeBrowse does not have this functionality but our analysis software VarSeq can perform this as long as your variants have been called and VCF files produced. Let me know if you have any further questions or if you would like to give VarSeq a try. Thanks, Jami... |
| 2015-09-24 15:54:26 -0600 | answered a question | how to get reference genome, recieving a message that says "Not enough bytes in packed blob to parse out data" Hi Russell, I wanted to followup from our phone conversation that downloading a new local copy of the reference sequence track was able to resolve the error message you were receiving. Please let me know if you have any further issues. Thanks, Jami.. |
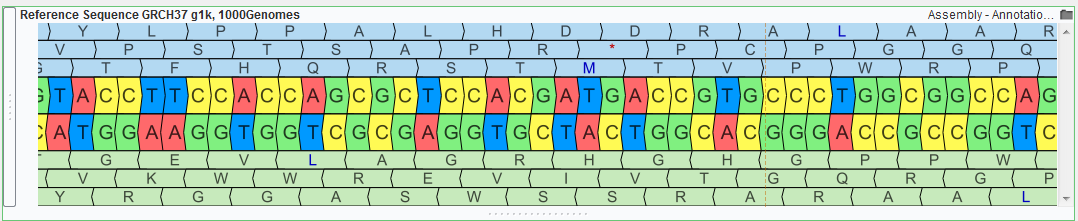
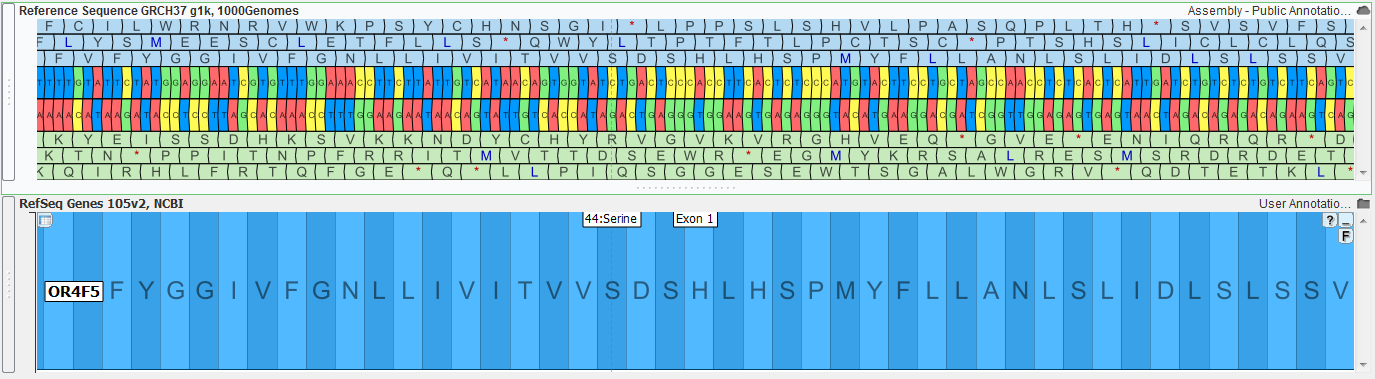
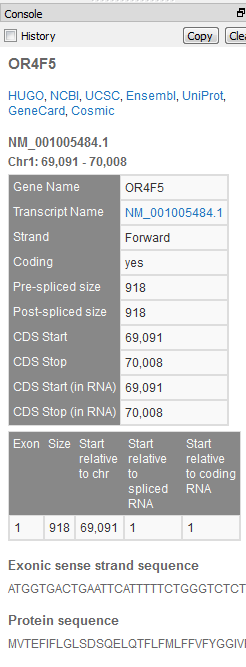
| 2015-08-17 12:02:42 -0600 | answered a question | amino acid information Dear Guy, I am happy to help with your GenomeBrowse questions. Zooming in on the select gene source to a base pair level will make the protein sequences visible, additionally the hover text for the exon region of the gene will contain the exon number. The below screenshot shows as zoomed in region (chr1: 69,162 - 69,296) of both the reference sequence and gene annotation source for the human GRCh37_g1k genome build.
Clicking anywhere in the exon region will provide further details for the gene in the Details pane.
Let me know if you have any further questions. Thanks, Jami... |
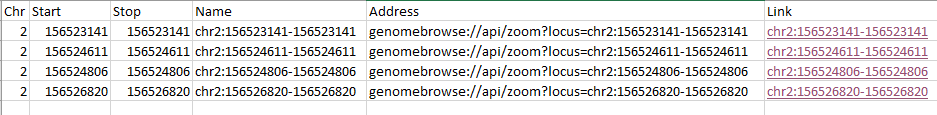
| 2015-07-20 11:45:13 -0600 | answered a question | Hyperlink to GenomeBrowse from another application? HI Siavosh, I am happy to help with your hyperlink questions. You can link to GenomeBrowse from third party software (ex. Excel, Evernote...) by using a simple html hyperlink similar to the URL: genomebrowse:/api/zoom?locus=chr1:476471220-47647338. Excel does not really like this URL format inside their HYPERLINK function so in the below example I used the CONCATENATE function to create the full strings before forming the hyperlinks.
So starting from a spreadsheet with at least chromosome, start and stop position you automatically create the hyperlinks by adding following additional columns.
If you click the links GenomeBrowse should open and zoom to that location or just perform the zoom if GenomeBrowse is already running. Let me know if you have any issues or further questions. Thanks, Jami... |
| 2015-04-27 09:22:07 -0600 | answered a question | Loading many files with a batch script Hi Olga, GenomeBrowse does not currently have batch script capabilities directly, so the only way to load a large number of files automatically is through the process of using the IGV functionality as you have described. GenomeBrowse does have that ability to drag and drop files into the plotting window. So another option would be to open a file explorer to the location of all of your BAM files and then drag and drop the ones you would like to visualize directly into the plot window. Depending on the size of the files you may want to use our command line program "gautil" to first compute index and coverage for the files so these computation does not overwhelm your system. See our manual for further details. Let me know if you have any further questions. Thanks, Jami... |
| 2015-04-27 09:12:02 -0600 | answered a question | Annotation tracks for pan troglodytes Hi Martin, As follow-up from our email discussion I wanted to see if you were able to successfully add the custom genome for Chimpanzee into GenomeBrowse? You can curate your own reference sequence by converting the FASTA reference that was used to align your sequences and create your BAM files. To do this go to Add and select Convert in the lower left corner, add your FASTA file and follow the prompts to create the reference sequence. You can find an example workflow for this process in a tutorial we have for our analytic software SVS at the following link, the process is very similar in GenomeBrowse. Create Custom Genome Assembly Once the conversion is complete you will want to close and reopen GenomeBrowse for the new source to be available, then select the new genome build from the assembly dropdown and your BAM files should correctly draw in GenomeBrowse. Alternatively we can also create the reference sequence and have it available from our Public Annotations repository. If you can provide a link to a public source of the references sequence I can add it to our list of genomes to curate. Let me know how I can help you further. Thanks, Jami… |
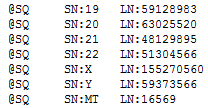
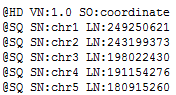
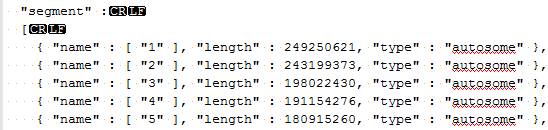
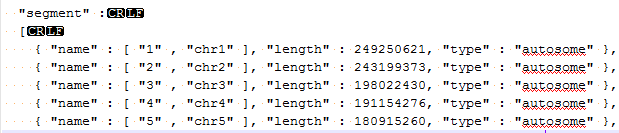
| 2015-04-27 09:11:10 -0600 | answered a question | error code 18 Hi Mike, I am sorry you are having issues loading your BAM files for visualization in GenomeBrowse. The "Error Code 18" message is an error that occurs when there is a mismatch between either the chromosome names or chromosome lengths that are listed in your converted reference sequence and the header lines of your BAM file. If you click on the BAM file in the Plot Tree and then scroll to the bottom of the Console window you can see the values that are listed in your BAM file, it should look similar to the following where the SN values are the chromosome names and the LN values are the lengths of each region.
This information needs to match what is listed in the genome assembly file that was created when you converted your FASTA file to a reference sequence source. If you go to Tools > Annotations Folder and then move up one folder level to the /CommonData/ folder you should see a folder called /Assemblies/, if you open the assembly file for your species and build in a text editor (Notepad++) you should see something like the following.
If these values are different (aside from the extra “chr” in the chromosome name) then you will see the error message you reported. If it is the chromosome names that are different you can add additional names directly to the assembly file, for example like the following:
Then just save your changes, close and reopen GenomeBrowse so it will recognize the changes and you should be good to go. If the length values are different then that is a more of an issue to change. If you can provided screenshots of what you are seeing I may be able to provide further suggestions. Alternatively if you would be willing to share your BAM and FASTA file I can take a look directly at the data to see what may be going wrong. Let me know how I can help you further. Thanks, Jami… |
| 2015-04-27 09:06:22 -0600 | answered a question | How to install GenomeBrowse on a cluster/server? Hi Cesar, As long as some type of GUI forwarding is set up from your server through your remote connection (we use MobaXterm with X11 forwarding to connect to our Linux machines) then GenomeBrowse should be able to be installed and used on your system. Each user will need to register a username and password for GenomeBrowse for access to the software. If you run into any issues while you are trying to install GenomeBrowse please let us know. Screenshots of error or console messages are most useful. Let me know how I can assist you further. Thanks, Jami… |
| 2015-04-27 09:04:28 -0600 | answered a question | converting gff files Unfortunately the GenomeBrowse Convert Wizard does not support conversion of GFF files. Do your GFF files contain gene annotation data? The Convert Wizard is only able to handle GTF format for gene annotation sources or a more generalized text based format where each gene is a single line in the text file. We have tools available in our analysis software, SVS, that can convert GFF files but since those tools are written in Python they are unable to be used in the stand-alone version of GenomeBrowse. What type of data are you trying to convert? If the data type can be supported in GTF format you may want to covert the GFF data directly to GTF then you will be able to use the Convert Wizard to reformat the data for loading in GenomeBrowse. Please let me know how I can help you further. Thanks, Jami… |
| 2015-04-27 08:59:34 -0600 | answered a question | connection refused Dear Dr. Boidot, I am sorry you are having issues logging in to GenomeBrowse. "Connection Refused" error messages are generally caused by something blocking GenomeBrowse from connecting to our servers. If your network requires a proxy to access outside sources then you will want to verify that the settings for your proxy have not changed. If they have changed then you will want to go to Tools > Proxy Settings to update GenomeBrowse with the most recent information. Please let me know if I can help you further. Thanks, Jami... |
| 2015-04-27 08:54:16 -0600 | answered a question | How can I load the OMIM data to GenomeBrowse? Thank you for your question. If you have the OMIM data available in a supported format (i.e., delimited text, VCF, BED, etc.) then that file can be converted to TSF format (GenomeBrowse format) using our Convert Wizard (File > Convert). http://doc.goldenhelix.com/GenomeBrowse/latest/gbmanual/convertsourcewizard.html Once converted to a supported format you can add the data for visualization through the Add dialog of your GenomeBrowse project. Please let me know if you have any further questions. Thanks, Jami... |
| 2015-01-16 09:40:07 -0600 | received badge | ● Enthusiast |
| 2014-10-17 11:47:47 -0600 | answered a question | Having trouble converting bigwig files. Hi Barbara, I am sorry you are having issues converting your files. Unfortunately we had to pull the functionality to convert BigWig and BigBed files from the Convert Wizard due to visualization issues with this data type. We forgot to remove these file types from the dropdown menu when we removed support for the data. So any BigWig or BigBed files added to the Convert Wizard will produce the error you have seen. Once we are able to add back in support for these file types we will let you know. Sorry of the inconvenience. Thanks, Jami... |
| 2014-10-17 11:47:05 -0600 | answered a question | Having trouble converting bigwig files. Hi Barbara, I am sorry you are having issues converting your files. Unfortunately we had to pull the functionality to convert BigWig and BigBed files from the Convert Wizard due to visualization issues with this data type. We forgot to remove these file types from the dropdown menu when we removed support for the data. So any BigWig or BigBed files added to the Convert Wizard will produce the error you have seen. Once we are able to add back in support for these file types we will let you know. Sorry of the inconvenience. Thanks, Jami... |
| 2014-09-04 11:29:38 -0600 | answered a question | GRCh38/Hg38 assembly Hi Kim, We have made the GRCh38 human build available in both GenomeBrowse and SVS. If you select the genome assembly drop-down at the top of the GenomeBrowse window and scroll to the Human assemblies you should see the four human assemblies that we currently have available.
The GRCh38 assembly was created from the data available directly from NCBI so should match up to the hg38 data that is available from UCSC. If you are getting an error message or your data is not correctly loading please let me know and we can look into the issue. Thanks, Jami... |
| 2014-08-19 11:14:50 -0600 | answered a question | custom reference : Unable to find reference sequence source for build Hi Michael, I was able to take a look at the GRCH37g1k fasta and it looks like our Convert Wizard has some issues with the GZ version of the file, when loading it into the Convert Wizard and after the scanning phase completes if you scroll to the bottom of the segment list, it seems to be picking up some strange encoding from the compression of the file.
If you load the unzipped file into the Convert Wizard the extra data at the bottom disappears and once you rename the FASTA segments to the standard 1,2,3,etc. this reference file seems to match up to the GRCh37_g1k TSF we have available. Let me know if you continue to have issues or if you have any further questions. Thanks, Jami... |
| 2014-08-14 10:49:08 -0600 | answered a question | custom reference : Unable to find reference sequence source for build Hi Michael, I am sorry you are also having issues with the gautil functionality. For precomputing BAM coverage using the "coverage" function of gautil, it will by default look for the reference sequence TSF file in the ../GenomeBrowse/Application/Data folder. If the reference file is saved in a different directory then you can use the --refFolder="folder location" command to point to the new location. As an example if you had your BAM and index files (sample.bam and sample.bam.bai) stored on a network drive (M) and the reference file was stored on an external hard drive (G) then the command would look something like the following. gautil coverage M:/sample.bam --refFolder="G:/" or ./gautil.exe coverage M:/sample.bam --refFolder="G:/" if you are on a Linux machine. The tool will then pick out the correct reference TSF file to use for computing the coverage based on the chromosome names and lengths listed in the header of your BAM file. The names and lengths listed in the BAM must match exactly to what is listed in the TSF file for the coverage tool to be able to identify the correct reference. Please let me know if you continue to have any issues. We will also look into improving the documentation for the gautil tool to be more comprehensive and accurate. Thanks, Jami... |
| 2014-07-15 15:25:07 -0600 | answered a question | Proxy Settings Hi Vicky, I am sorry you are having issues accessing our data servers to download annotation tracks. If you are running a firewall that must have explicit allow rules for programs to make outgoing connections you need to allow both "GoldenHelix.exe" and "aria2c.exe" to make HTTP and HTTPS outgoing connections (port 80 and port 443 respectively). The default Windows firewall does not require this but third party ones like F-Secure may be configured to require this. If this is not causing the issue can you provide details on the exact error message you are receiving? Let me know how I can assist you further. Thanks, Jami... |
| 2014-07-10 16:46:42 -0600 | answered a question | Is there a downloadable annotation file from ENCODE to map my SNP? Hi Philip, We do not currently have an annotation source for Encode data. The data available from this source is provide in BigBed and BigWig formats (here) and we are still working on implementing reading these file types into GenomeBrowse. The Encode annotation database is on our list of annotation sources to be made available and as soon as our BigBed and BigWig functionality is ready we will have the track created and uploaded to our servers. I will let you know as soon as it becomes available. Let me know if you have any further questions. Thanks, Jami... |
| 2014-07-01 08:44:14 -0600 | answered a question | custom reference : Unable to find reference sequence source for build Hi Yadhu, I am sorry you are having issues computing the coverage for your BAM file. The gautil function for coverage computations does not directly support using the FASTA formatted reference sequence. The reference file must be converted to Golden Helix TSF format using the Convert Data Wizard as a first step before it can be used in conjunction with gautil functions. If you launch GenomeBrowse and open the Add dialog you can click Convert... in the lower left corner of the Data Source Library to launch the converter. You would then add your FASTA file and follow the prompts to create the TSF reference sequence file. You can find specifics on the options for converting in our manual at the following link. http://doc.goldenhelix.com/GenomeBrowse/2.0.2/convertsourcewizard.html Please let us know if you have any further issues. Thanks, Jami... |
| 2014-06-09 11:42:16 -0600 | answered a question | VCF file support Hi Tamar, I am sorry you are having issues loading your BAM and VCF files into GenomeBrowse. Have you tried loading them through the Add dialog by navigating to their saved location? Our records indicate that you are using the Win64 version, if that is correct there is a know bug in that version for visualizing BAM files which may be causing the issue. Can you try updating to GenomeBrowse 2.0.2 to see if that resolves the issue. You can download the installer from our website at the following link. http://www.goldenhelix.com/GenomeBrowse/index.html If you are still seeing the same issues after updating could provide some screenshots of the error message, or even provide a sample of the data so we can try and reproduce the issue. You can email me directly at genomebrowse@goldenhelix.com to share your data in a secure fashion. Please let me know how I can be of further assistance. Thanks, Jami... |
| 2014-06-09 11:31:14 -0600 | answered a question | Crash when loading a BAM file in GenomeBrowse Win64 2.0.1 Hi Zhuo, I wanted to let you know that we have released GenomeBrowse 2.0.2. With the new version we have fixed the bug that was causing the Windows 64bit version of GenomeBrowse to crash when loading BAM files for visualization. You can update to the new version by downloading the installer from our website. http://www.goldenhelix.com/GenomeBrowse/index.html Please let us know if you have any further issues. Thanks, Jami... |
| 2014-05-29 16:17:51 -0600 | answered a question | Converting fasta for reference sequence Hi Krystel, I am sorry you are having issues creating your reference sequence track. Our Data Convert Wizard is very specific in the supported file formats and extensions for each type of conversion. In particular, for the FASTA converter the files must be *.fa or *.fasta or the *.gz versions for the wizard to pick the correct convert options. If you are downloading your reference sequence from the NCBI FTP site for the Enterobacteria phage lambda virus genome then the file with the closest supported format is NC_001416.fna. Before adding the file to the converter you will first want to rename the extension to be *.fa.
Then on the next dialog you will need to rename the segments (chromosome/scaffolds) present in the file since NCBI files contain more than just chromosome names in the headers for their FASTA files. Additionally if you have BAM files for this species then you will want to make sure the segment names you select for the convert wizard match exactly to what is contained in the header of the BAM file. In the below screenshot I have chosen to rename the segment to "1" and then listed the NCBI naming convention "NC_001416.1" as the Alias name. Either the "Segment" or "Alias Name" can be used to match the BAM file naming convention.
Also at this point you will want to create a Build Name for your genome. We generally use the assembly name given by NCBI, but for this genome an assembly name does not exist so you can just give it an informative name that you will recognize, keeping in mind that if you create any further data sources for this genome you will want to use the same naming convention. If you continue to have issues creating and using the reference sequence for this genome please let me know. If you could provide a link to the file you are trying to convert or provide a copy of the file to me at genomebrowse@goldenhelix.com that would be most useful. Thanks, Jami... |
| 2014-05-14 13:36:47 -0600 | answered a question | Other genomes? (Specifically Felis catus) Hi Brian, I wanted to let you know that the Felis catus (felCat5/Felis_catus-6.2) genome is now available for download from the Public Annotations location of the Add Dialog. Please let me know if you have any additional requests or further questions. Thanks, Jami... |
| 2014-05-09 09:13:14 -0600 | answered a question | Crash when loading a BAM file in GenomeBrowse Win64 2.0.1 Dear Zhuo, We have identified a bug in the Windows 64bit version of GenomeBrowse 2.0.1, the issue occurs when trying to create the index (BAI) and coverage (COVTSF) files that are necessary for visualization of BAM data. If you only have your BAM file in the directory you will see an error message "Error reading BAM: Failed bgzf_check_EOF (error code 3)", if you already have a BAI file in addition to your BAM then GenomeBrowse will crash when trying to compute the coverage file. We are working to fix this issue and should have an update available within the next few weeks. In the meantime you can download the Windows 32bit version of GenomeBrowse to use for computing the index and coverage files. Please let me know if you have any further issues or any questions. Thanks, Jami... |
| 2014-05-02 16:15:07 -0600 | answered a question | Importing hg18 reference genome Hi Danny, GenomeBrowse can support reference sequences for any species or build as long as there is reference allele FASTA file available for that genome assembly. For the human hg18 (NCBI36) build we already have a reference sequence created and available for download from our Public Annotations servers. You go to File > Add and select the Public Annotations tab and then select to show all human allele sequence tracks, you should see "Reference Sequence NCBI36, UCSC" track available for download. If you are using this build to load BAM files then you will need to make sure the chromosome names and lengths used to the header of the BAM file match exactly what is in the Genome Assembly file for this build. You can find the assembly file by going to Tools > Program Folder and inside the GenomeMaps folder will be text assembly file that lists the specifics for this build. Let me know if you have any further questions. Thanks, Jami... |
| 2014-04-30 11:40:37 -0600 | answered a question | Other genomes? (Specifically Felis catus) Hi Brian, We have the Felis catus (felCat5) genome on our list of genomes to support but have not had the chance to get it completed yet. It is on the top of my list and should be available by the beginning of next week. In the meantime if you are using GenomeBrowse 2.0 you can create the genome yourself if you have a reference allele FASTA file for the species. If you go to File > Convert... this will launch the convert wizard to assist you with adding the genome. If you have any questions click the Help button on the wizard to open the GenomeBrowse manual. Please let me know if you have further questions or issues. Thanks, Jami... |