
| 2017-11-09 15:39:10 -0600 | answered a question | How can I exclude an custom assembly? Hi Tiago, You will want to remove the assembly file found in your user data folder. To access this folder from GenomeBrowse, you can click tools > Open Folder > Annotations folder. Then back-up one directory to find the Assemblies folder. You can remove the custom assembly here. |
| 2017-11-09 15:36:49 -0600 | commented answer | Stranded Paired-end RNA-seq coloring and wig/bw support Also, UCSC offers some tools to make the conversions between these formats. http://genome.ucsc.edu/goldenpath/help/bigWig.html |
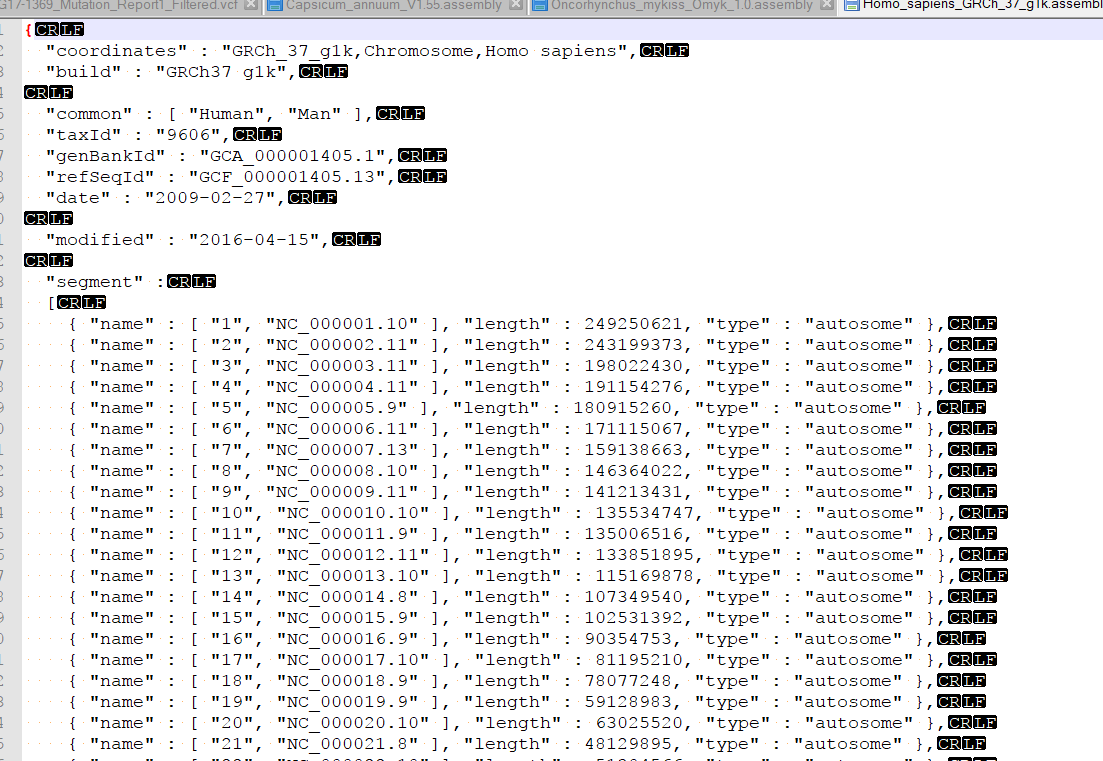
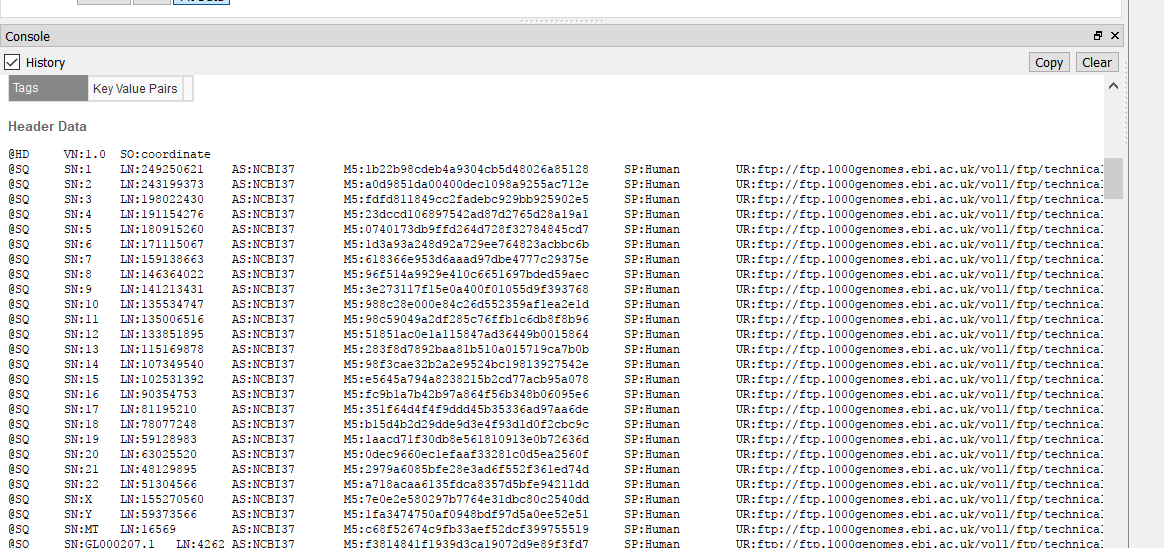
| 2017-11-09 15:17:39 -0600 | answered a question | Unable to Match Data in this Source with Reference Hi Brian, This sounds like there is a discrepancy between the headers in your BAM file and the names of the scaffolds in the assembly file that is created when converting your genome fasta in GB. You could check by plotting the BAM in GB and under the console window, examining the coordinates in the BAM file. Then compare these coordinates to what is found in the assembly file. This is typically found in your user data folder. To access this folder from GenomeBrowse, you can click tools > Open Folder > Annotations folder. Then back-up one directory to find the Assemblies folder. you can open the assemblies file in a text editor and change the order of the chromosomes.
|
| 2017-11-09 15:07:45 -0600 | commented answer | How to sort chromosomes in Golden Helix Genome Browser? FYI if you are interested in SVS (which has GenomeBrowse built-in) we can curate these assemblies for free! |
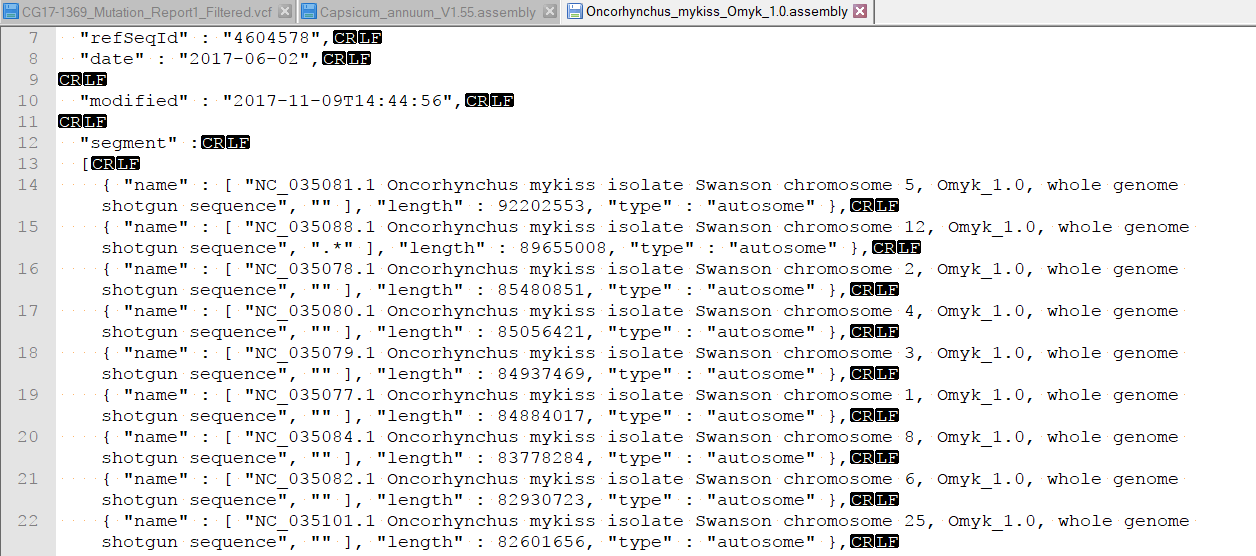
| 2017-11-09 15:06:56 -0600 | answered a question | How to sort chromosomes in Golden Helix Genome Browser? Hi Shawn, Was it the new Oncorhynchus mykiss (assembly Omyk_1.0) ? If you add a FASTA file from a new assembly to Genomebrowse, an assembly file gets created which specifies the order of the chromosomes listed. By default these chromosomes are sorted by size and not by number. If you want to edit the order in which these are displayed, you will have to edit the assembly file. This is typically found in your user data folder. To access this folder from GenomeBrowse, you can click tools > Open Folder > Annotations folder. Then back-up one directory to find the Assemblies folder. you can open the assemblies file in a text editor and change the order of the chromosomes.
|
| 2017-09-21 11:42:46 -0600 | answered a question | how to visualise sorted bam files? Hi Katerina, This typically means GB is computing a genomic index and the view will be available once the computation is complete. can you try loading a single BAM, zooming all the way in? |
| 2017-08-17 10:30:40 -0600 | received badge | ● Enthusiast |
| 2017-08-09 12:03:05 -0600 | received badge | ● Supporter (source) |
| 2017-08-09 12:02:13 -0600 | answered a question | READ DEPTH TRESHOULD Hi Rute, For Sanger sequencing typically you would look at the Chromatograms. Here is a nice webpage that provides additional details. https://seqcore.brcf.med.umich.edu/si... If you goal is to view a chromatogram to view ab files and assess SNP calls from Sanger sequencing, there are a variety of free tools (genestudio, Sequence Scanner, 4 peaks, etc…). Kind Regards, -Steve |
| 2017-08-09 11:47:57 -0600 | answered a question | No E coli BW25113 genome? Hi Arya, Unfortunately we do not currently support the E.coli BW25113 genome assembly. Typically, an allele reference sequence source can be built for any species where there is an available DNA sequence (FASTA) file. The Convert Wizard that is available within the software can directly support conversion of the GTF formatted data. The Convert Wizard can be accessed by going to File > Add then clicking Convert in the lower left corner of the Data Source Library. Once the FASTA data is converted to TSF (Golden Helix annotation format) then it can be loaded into the GenomeBrowse plot window for visualization. Let me know if you have any further questions. Best, -Steve |
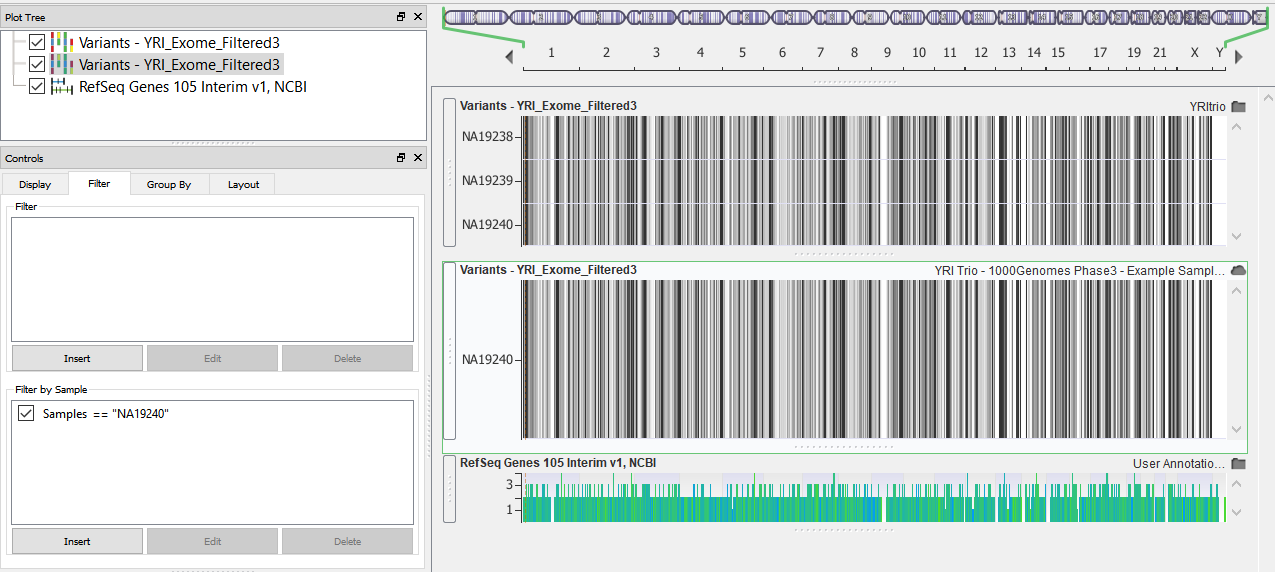
| 2017-08-08 16:45:45 -0600 | answered a question | Is there a way to view only a single sample in a multi-vcf file Yes. In the controls view you can add a filter to examine a single sample in a multi-sample VCF. The expression Samples =="sample name" should do the trick. |
| 2016-11-29 10:33:57 -0600 | answered a question | Can you export sequence information? Cassidy, Are you looking to export sequence from a specific gene from the available reference sequence? If so, first you need to plot the reference sequence by clicking on the plot icon in the tool bar and selecting which build. Once the reference sequence is displayed you can zoom into your area of interest >right click and select 'Feature List'. Here is where you are going to find sequence data. From here you can highlight all (ctrl+A) or select a subset of the rows and right click > copy selected sequence to clipboard. A good rule of thumb, the feature list will only capture the sequence you have displayed. So for more sequence information, simply zoom out and select 'Feature List' . For less sequence zoom in and select 'Feature List'. Let me know if this answers your question, -Steve |
{kind=link}