Hi Mike,
I am sorry you are having issues loading your BAM files for visualization in GenomeBrowse.
The "Error Code 18" message is an error that occurs when there is a mismatch between either the chromosome names or chromosome lengths that are listed in your converted reference sequence and the header lines of your BAM file.
If you click on the BAM file in the Plot Tree and then scroll to the bottom of the Console window you can see the values that are listed in your BAM file, it should look similar to the following where the SN values are the chromosome names and the LN values are the lengths of each region.
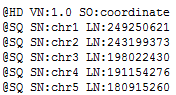
This information needs to match what is listed in the genome assembly file that was created when you converted your FASTA file to a reference sequence source. If you go to Tools > Annotations Folder and then move up one folder level to the /CommonData/ folder you should see a folder called /Assemblies/, if you open the assembly file for your species and build in a text editor (Notepad++) you should see something like the following.
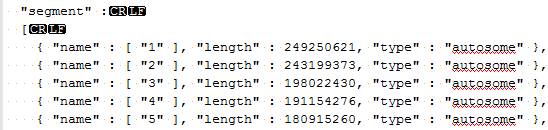
If these values are different (aside from the extra “chr” in the chromosome name) then you will see the error message you reported. If it is the chromosome names that are different you can add additional names directly to the assembly file, for example like the following:
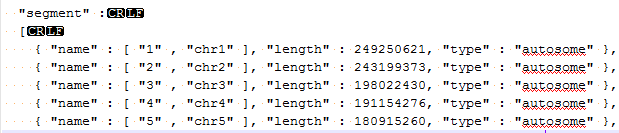
Then just save your changes, close and reopen GenomeBrowse so it will recognize the changes and you should be good to go. If the length values are different then that is a more of an issue to change.
If you can provided screenshots of what you are seeing I may be able to provide further suggestions. Alternatively if you would be willing to share your BAM and FASTA file I can take a look directly at the data to see what may be going wrong.
Let me know how I can help you further.
Thanks,
Jami…
